Sequencing variability
Abstract
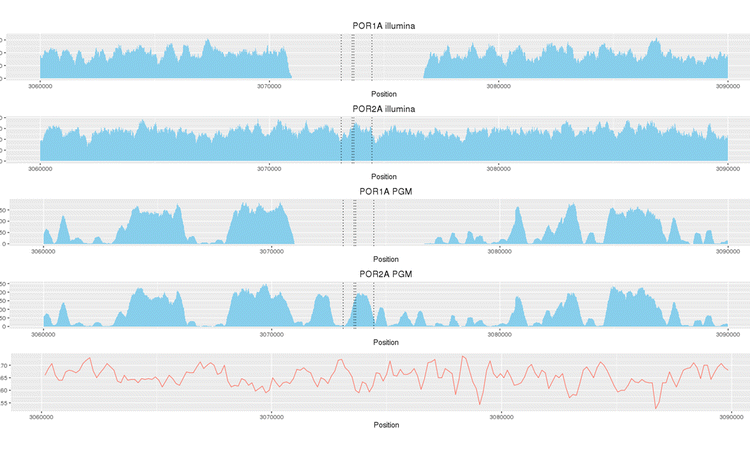
The emergence of resistance to anti-tuberculosis drugs is a serious and growing threat to public health. Next-generation sequencing is rapidly gaining traction as a diagnostic tool for investigating drug resistance in Mycobacterium tuberculosis to aid treatment decisions. However, there are few little data regarding the precision of such sequencing for assigning resistance profiles. We investigated two sequencing platforms (Illumina MiSeq, Ion Torrent PGM™) and two rapid analytic pipelines (TBProfiler, Mykrobe predictor) using a well characterised reference strain (H37Rv) and clinical isolates from patients with tuberculosis resistant to up to 13 drugs. Results were compared to phenotypic drug susceptibility testing. To assess analytical robustness individual DNA samples were subjected to repeated sequencing. The MiSeq and Ion PGM systems accurately predicted drug-resistance profiles and there was high reproducibility between biological and technical sample replicates. Estimated variant error rates were low (MiSeq 1 per 77 kbp, Ion PGM 1 per 41 kbp) and genomic coverage high (MiSeq 51-fold, Ion PGM 53-fold). MiSeq provided superior coverage in GC-rich regions, which translated into incremental detection of putative genotypic drug-specific resistance, including for resistance to para-aminosalicylic acid and pyrazinamide. The TBProfiler bioinformatics pipeline was concordant with reported phenotypic susceptibility for all drugs tested except pyrazinamide and para-aminosalicylic acid, with an overall concordance of 95.3%. When using the Mykrobe predictor concordance with phenotypic testing was 73.6%. We have demonstrated high comparative reproducibility of two sequencing platforms, and high predictive ability of the TBProfiler mutation library and analytical pipeline, when profiling resistance to first- and second-line anti-tuberculosis drugs. However, platform-specific variability in coverage of some genome regions may have implications for predicting resistance to specific drugs. These findings may have implications for future clinical practice and thus deserve further scrutiny, set within larger studies and using updated mutation libraries.
Figures
Citation
Phelan, J., O’Sullivan, D. M., Machado, D., Ramos, J., Whale, A. S., O’Grady, J., … Clark, T. G. (2016). The variability and reproducibility of whole genome sequencing technology for detecting resistance to anti-tuberculous drugs. Genome Medicine, 8(1), 132