Protein structural modelling
Abstract
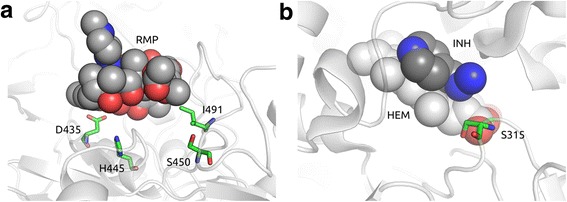
Combating the spread of drug resistant tuberculosis is a global health priority. Whole genome association studies are being applied to identify genetic determinants of resistance to anti-tuberculosis drugs. Protein structure and interaction modelling are used to understand the functional effects of putative mutations and provide insight into the molecular mechanisms leading to resistance. To investigate the potential utility of these approaches, we analysed the genomes of 144 Mycobacterium tuberculosis clinical isolates from The Special Programme for Research and Training in Tropical Diseases (TDR) collection sourced from 20 countries in four continents. A genome-wide approach was applied to 127 isolates to identify polymorphisms associated with minimum inhibitory concentrations for first-line anti-tuberculosis drugs. In addition, the effect of identified candidate mutations on protein stability and interactions was assessed quantitatively with well-established computational methods. The analysis revealed that mutations in the genes rpoB (rifampicin), katG (isoniazid), inhA-promoter (isoniazid), rpsL (streptomycin) and embB (ethambutol) were responsible for the majority of resistance observed. A subset of the mutations identified in rpoB and katG were predicted to affect protein stability. Further, a strong direct correlation was observed between the minimum inhibitory concentration values and the distance of the mutated residues in the three-dimensional structures of rpoB and katG to their respective drugs binding sites. Using the TDR resource, we demonstrate the usefulness of whole genome association and convergent evolution approaches to detect known and potentially novel mutations associated with drug resistance. Further, protein structural modelling could provide a means of predicting the impact of polymorphisms on drug efficacy in the absence of phenotypic data. These approaches could ultimately lead to novel resistance mutations to improve the design of tuberculosis control measures, such as diagnostics, and inform patient management.
Figures
Citation
Phelan, J., Coll, F., McNerney, R., Ascher, D. B., Pires, D. E. V, Furnham, N., … Clark, T. G. (2016). Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti-tuberculosis drug resistance. BMC Medicine, 14(1), 31