GWAS lecture
Genome Wide Aassociation Studies (GWAS) Practical
The objective of this tutorial is to get you familiar with the basic file format used for GWAS and common tools used for analysis and take you through data Quality control (Crucial in any study!).
Our dataset is based on a GWAS study for Meningococcal disease in a European population (https://www.nature.com/articles/ng.640).
Data:- Genome wide SNP data
- Scripts to facilitate analysis
- Computer workstation with Unix/Linux operating system
- PLINK software for genome-wide association analysis: http://pngu.mgh.harvard.edu/_purcell/plink/download.shtml
- Statistical software for data analysis and graphing such as: R: http://cran.r-project.org/
- SMARTPCA.pl software for running principal components analysis (not used in this tutorial to save time): http://genepath.med.harvard.edu/~reich/Software.htm
- BCFtools
conda environment. Activate it with:
conda activate gwas
A. Create BED files for analysis:
1. Convert your plink genotype files to binary format - smaller file easier for manipulation of data
We have already provided you with plink formatted files we won't have to do this step.
plink allows for the conversion from many different formats to its own format. For example if you had
a VCF file you
could type:plink --vcf MD.vcf.gz --make-bed --out MD
Your data set:
A plink-binary-formatted dataset consisting of 3004 individuals, 409 cases, 2595 controls, 601089
variants. The three files starting with MD. hold all the information plink needs:
MD.bed – binary-encoded information on individuals and variantsMD.bim – variant information: “Chromosome”, “Marker name”, “Genetic Distance” (or '0' as dummy
variable),
“Base-pair coordinate”, “Allele 1”, “Allele 2”. Each SNP must have only two alleles.MD.fam – information on the individuals: The first 6 columns are mandatory and in the order: “Family
ID”, “Individual ID”,
“Paternal ID”, “Maternal ID”, “Sex”, “Phenotype”.Double-check the basic stats of your dataset (number of variants, individuals, controls, cases) by examining MD.bim and MD.fam with bash utilities like awk or wc.
MD.fam holds one line per individual and MD.bim has one line per variant. We can
therefore use the word-counting utility wc to get those numbers with
wc -l MD.{bim,fam}
Alternatively, you can count the number of lines with awk like so
awk 'END{print NR}' MD.bim
awk 'END{print NR}' MD.fam
In order to get the number of controls and cases ('1' and '2' in the 6th column in the .fam
file,
respectively), we can again use awk, which is very versatile:
awk 'BEGIN{contr=0;cases=0;FS=" "}
{if($6==1){contr++} if($6==2){cases++}}
END{print "control", contr; print "cases", cases}' MD.fam
However, awk's syntax can be quite intimidating to beginners. Instead, we can get the
numbers of
cases and controls by chaining together a few other programs like so:
cut -f 6 -d ' ' MD.fam | sort | uniq -cThis gets the 6th column of the
MD.fam file and
counts the
occurrences of the unique values.
uniq expects sorted input. Try the command again without sort and see how the
output changes.
B. Sample QC
1. Identification of Individuals with discordant sex information
Ideally, if X-chromosome data are available, we would calculate the mean homozygosity
rate across X chromosome markers for each individual in the study and identify discordance
with our reported sex phenotype.
As our data only contains autosomes we will skip this step.
2. Identification of individuals with elevated missing data rates or outlying heterozygosity rate
2.1. At the shell prompt type:
plink --bfile MD --missing --out MD
This creates the files MD.imiss (sample-based missing report) and MD.lmiss
(variant-based missing report).
The fourth column in the .imiss file (N_MISS) gives the number of missing SNPs and the sixth column
(F_MISS)
gives the proportion of missing SNPs per individual.
2.2. At the shell prompt type:
plink --bfile MD --het --out MDThis creates the file MD.het where the third column gives the observed number of homozygous
genotypes [O(Hom)]
and the fifth column gives the number of non-missing genotypes [N(NM)], per individual.
2.3. Calculate the observed heterozygosity rate per individual using the formula (N(NM) - O(Hom))/N(NM) and
create a graph where the proportion
of missing SNPs per individual is plotted on the x-axis and the observed heterozygosity rate per individual is
plotted on the y-axis. We have provided an R script for this task. Run it by typing:
R CMD BATCH imiss-vs-het.Rscript
This creates the graph MD.imiss-vs-het.pdf (see below).
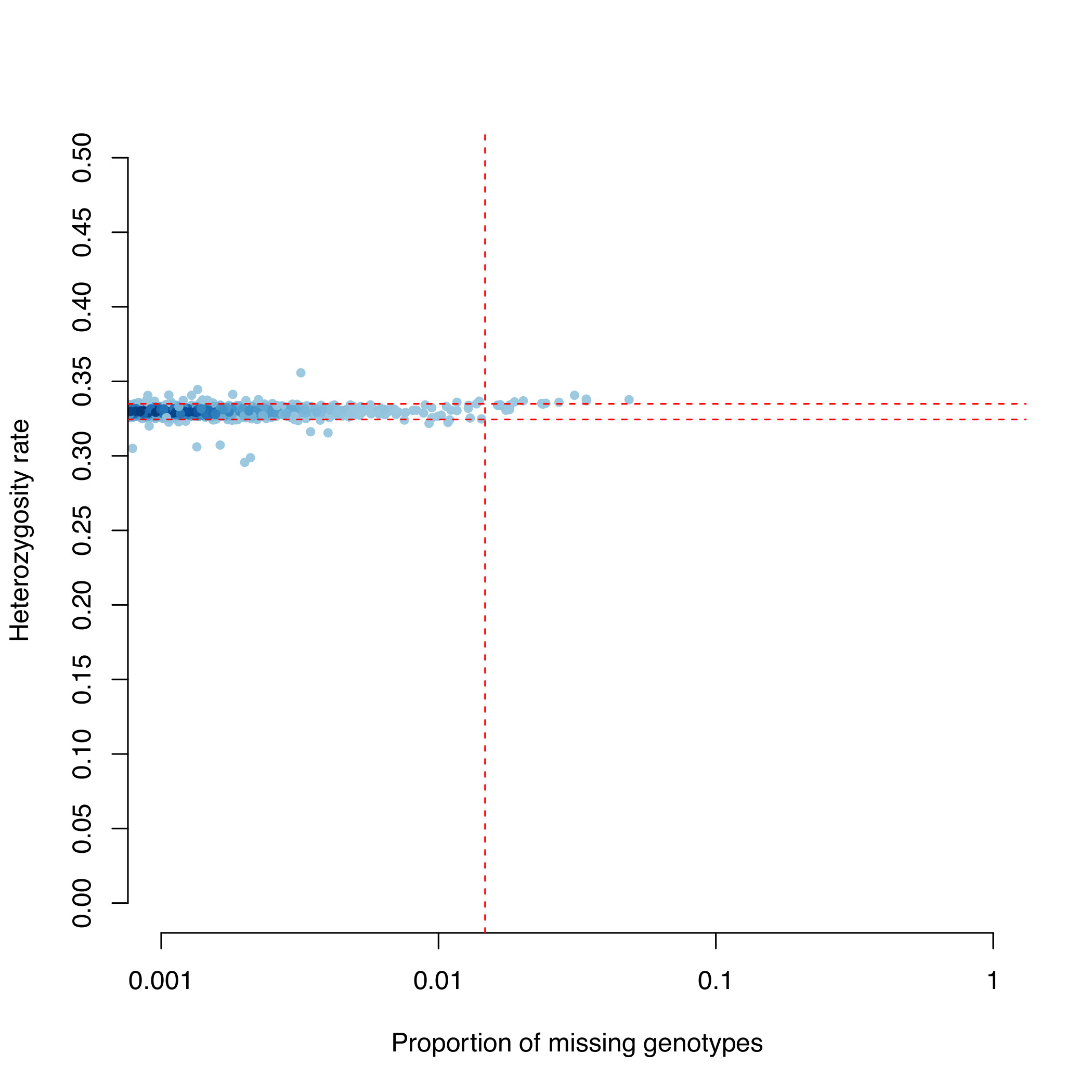
- genotype failure rate ≥ 0.0185 (vertical dashed line) and/or
- a heterozygosity rate ± 3 standard deviations from the mean (horizontal dashed lines).
2.4. Extract the family ID and individual ID of all samples failing this QC using another R script:
R CMD BATCH imiss_het_fail.Rscript
This produces a file named fail_imiss_het_qc.txt.
3. Identification of duplicated or related individuals
3.1. To identify duplicates & related individuals, create an identity-by-state (IBS) matrix – calculated for each pair of individuals based on the shared proportion of alleles.
3.1.1. To reduce the computational complexity, first prune the dataset so that no pair of SNPs (within a given window e.g 200kb) has linkage disequilibrium (r² > 0.2). Type
plink --bfile MD --indep-pairwise 200 5 0.5 --out MD
This creates files with the extensions .prune.in, .prune.out, and .log.
3.1.2. Then, to extract the pruned SNPs and generate the pair-wise IBS matrix, type:
plink --bfile MD --extract MD.prune.in --genome --out MD
This might take a few minutes and then creates files with the extensions .genome & .log.
3.1.3. We have provided a Perl script in order to identify all pairs of individuals with an
Idenity-by-descent (IBD) > 0.185. Invoke it with
perl run-IBD-QC.pl MDTo visualise the IBD rates, type:
R CMD BATCH plot-IBD.Rscript
this generates MD.IBD-hist.pdf:
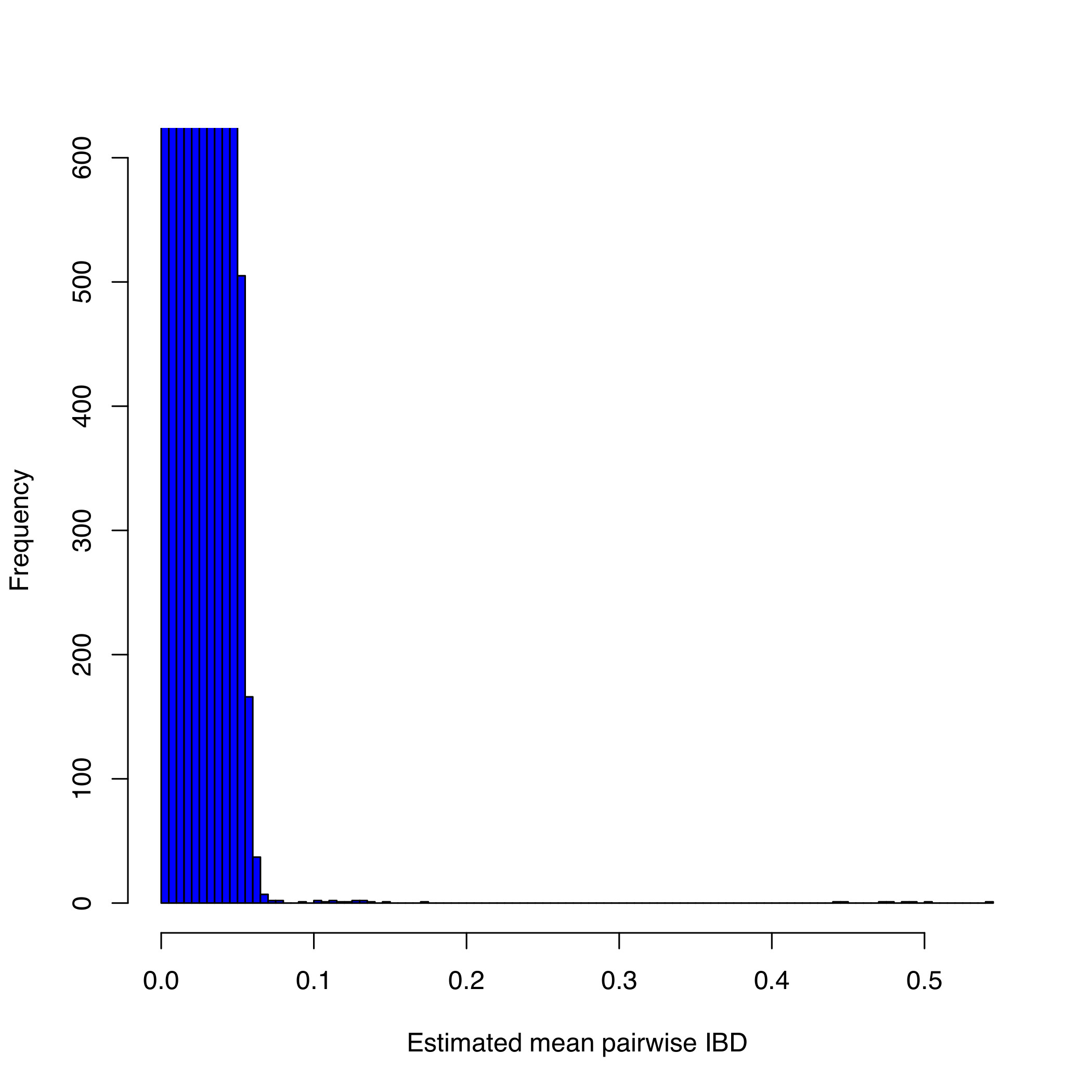
4. Identification of individuals of divergent ancestry
4.1. Principal components analysis (PCA) was performed with pruned bed file datasets generated from step 3.1.2
using the ./RUN_PCA.sh command. This generated the following output files (stored in
PCA_files): MD.pruned.pca.par,
MD.pruned.pca.log, MD.pruned.pca.evec, MD.pruned.pca,
MD.pruned.eval. The file with the .evec extension file is what you will
need to view your PCs.
4.2. Create a scatter diagram of the first two principal components, including all individuals in
PCA_files/MD.pruned.pca.evec (the first and second PCs are columns 2 and 3 respectively), using yet
another R script. Type:
R CMD BATCH plot-pca-results.Rscript
This creates pca_plot.pdf:
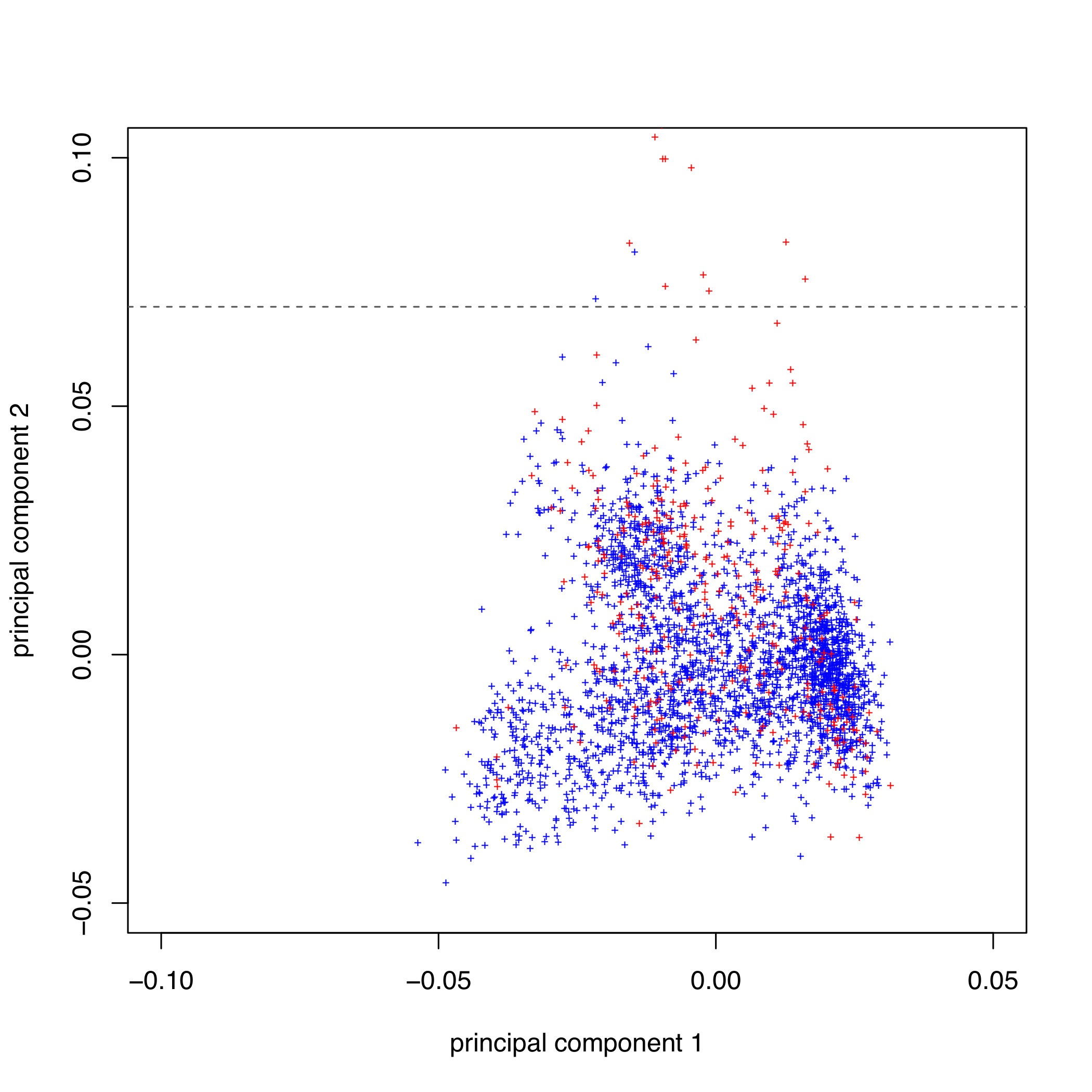
Next we can run
R CMD BATCH write_pca_fail.Rfail_pca.txt.
4.3. Remove all individuals failing QC
4.3.1. We can use the cat command to concatenate all the files listing individuals failing the previous QC steps into single file:
cat fail*txt | sort -k1 | uniq > fail_qc_inds.txt
The file fail_qc_inds.txt should now contain a list of unique individuals failing the previous QC
steps.
4.3.2. To remove these from the dataset type:
plink --bfile MD --remove fail_qc_inds.txt --make-bed --out clean.MDHow many individuals in total will be excluded from further analysis?
How many individuals in total do you have for further analysis?
clean.MD.fam.
Alternatively, you can use less clean.MD.log to have a look at the log-file of the previous step
and extract the information from there. This should tell us that we have 2888 samples left which means that
116 were removed.
C. Marker QC
5. Identify all markers with an excessive missing data rate
5.1. To calculate the missing genotype rate for each marker type:
plink --bfile clean.MD --missing --out clean.MD
The results of this analysis can be found in clean.MD.lmiss.
5.2. Plot a histogram of the missing genotype rate to identify a threshold for extreme genotype failure rate. This
can be done using the data in column five of the clean.MD.lmiss file. Use an R script provided for
this task by typing:
R CMD BATCH lmiss-hist.Rscript
This generates clean.MD.lmiss.pdf
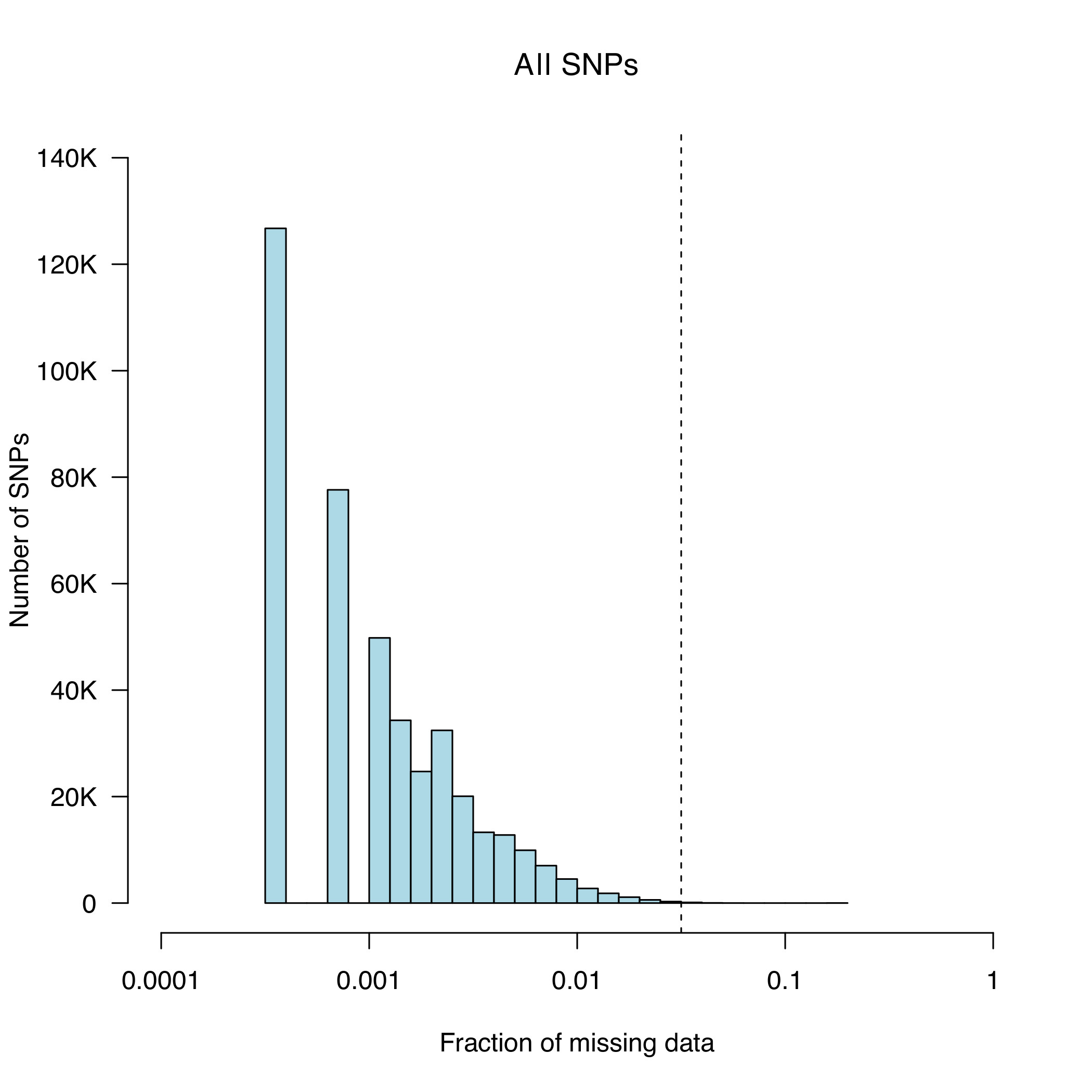
We chose to a call-rate threshold of 5% (these SNPs will be removed later in the protocol).
6. Test markers for different genotype call rates between cases and contols
6.1. To test all markers for differences in call rate between cases and controls, at the Unix prompt type:
plink --bfile clean.MD --test-missing --allow-no-sex --out clean.MD
The output of this test can be found in clean.MD.missing.
To create a file called fail-diffmiss-qc.txt, which contains all SNPs with a significantly different
(P<0.00001) missing rate between cases and controls, type
perl run-diffmiss-qc.pl clean.MD7. Remove all markers failing QC
7.1. To remove poor SNPs from further analysis and create a new clean (QC'D) MD data file, at the Unix prompt type:
plink --bfile clean.MD --exclude fail-diffmiss-qc.txt --maf 0.01 --geno 0.05 --hwe 0.00001 --make-bed --out clean.final.MDIn addition to markers failing previous QC steps, those with a MAF < 0.01, missing rate > 0.05 and a HWE P-value < 0.00001 (in controls) are also removed.
clean.final.MD.log. Towards the end of the file you will find the following
line:
8. Perform a GWAS on your QC'ed dataset
8.1. To run a basic case/control association test, at the unix prompt type:
plink --bfile clean.final.MD --assoc --ci 0.95 --adjust --allow-no-sex --out final.MD.assocYour association output file will contain 12 columns:
8.2. To visualise your data:
We will use another R script to generate two more plots:
8.2.1. A Quantile-Quantile (QQ) plot of your p-values to look at the distribution of P-values and assess
whether genomic inflation is present (lambda>1) (this can also be found in your .assoc.log file).
8.2.2. A Manhattan plot to visualise where your association signals lie across the chromosomes. Type:
R CMD BATCH GWAS_plots.R
This generates both plots: final.MD.assoc_qq.png and final.MD.assoc_mhplot.png
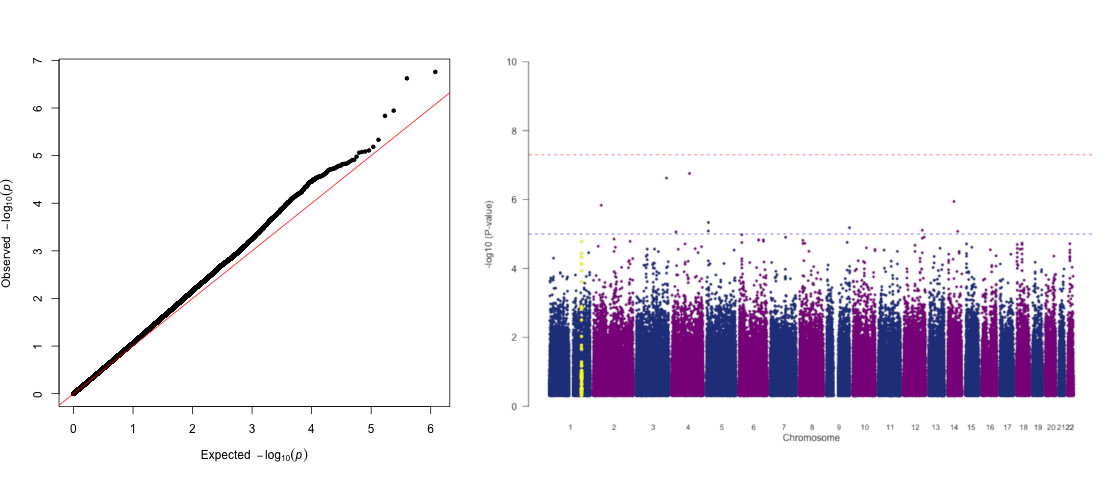
8.2.2.1. Let's zoom into a region of interest: the tower of SNPs on CHR1 (coloured in yellow). This the Complement
Factor H (CFH) region known to be associated with Meningococcal disease.
The previous Rscript in 8.2.2 above also generated the chr1_CFH_region.txt file.
- Open the locuszoom webpage: http://locuszoom.org/genform.php?type=yourdata
- Upload the text file and select PLINK data format.
- Enter the most associated snp (“rs1065489”) with a flanking size of 500KB
- In the Genome Build/LD Population field select the appropriate hg19 european ref panel.
- Then press "Plot Data" to generate your plot.